Even before I saw the scratchy old movie clips of Papua New Guineans afflicted with kuru shaking spasmodically in the days leading up to their inevitable deaths, I found prions frightening. There is something otherworldly about prions, akin to a horrific sci-fi plague or biblical scourge, and they seem to represent something that is living yet unliving. Unlike bacteria, viruses, fungi, parasites or any other disease agents, prions have no RNA or DNA, yet their simple misfolded protein structures allow them to behave like other fatal diseases. We tend to anthropomorphize regular diseases and describe them as evil, presumably because we view them as living entities that want to thrive at our expense. Prions however are like heartless automatons that just kill us in a methodical, cold-blooded manner. It gets a person wondering, what exactly constitutes life? The science explaining prions is still being debated, but the results are unquestionable – prions replicate inside mammalian brain tissue until the host animal dies, and the process is untreatable.
In this article I will describe the more common prion diseases, and explain our current understanding of how they work. These diseases usually go under the name of transmissible spongiform encephalopathy (TSE), and the three common varieties I will cover are kuru, bovine spongiform encephalopathy (BSE), known as mad cow disease, and Creutzfeldt-Jakob disease. Note that there are many other known TSE’s such as Gerstmann–Sträussler–Scheinker syndrome, Alpers’ disease and Fatal Familial Insomnia which are (rarely) found in humans, scrapie which affects sheep and goats, and chronic wasting disease which afflicts deer, elk, moose and other ungulates.
Replication
The sequence of events involved in prion diseases is fairly easy to describe. Mammals have a naturally occurring protein found abundantly in neural tissue throughout the body, but most importantly in the brain; this protein is given the name prion, or PrP. Its normal, properly folded state is denoted by the term PrPC with the ‘C’ superscript standing for ‘cellular’ or ‘common’. What purposes PrP serves is actively debated, but it appears to be used by the body for many different functions with evidence linking it to cell-to-cell communication, long term memory, and creation of bone marrow. The improperly folded forms which cause the diseases are generally given the term PrPSc with the ‘Sc’ referring to scrapie, which was the first form of prion disease to be identified by science. The PrP structure differs slightly from species to species, and hence the forms of PrPSc and the diseases they cause also vary between species, and sometimes the ‘Sc’ superscript is changed to indicate the specific variety of TSE.
If a dangerous (to that species) form of PrPSc is present in the body, getting there either through inheritance or some other way (e.g. ingestion or spontaneous mutation), then the PrPSc causes healthy PrP to transform into unhealthy PrPSc. This process becomes a runaway exponential chain reaction, with the converted prions forming insoluble fibroid amyloid aggregates which ultimately consume and replace healthy neural tissue, causing massive irreversible damage to the brain. The word spongiform is used because it describes the spongy porous look of brain tissue damaged by TSE diseases (Fig. 1). Note that the vacuoles created can be up to 70mm wide in advanced cases, such as that shown in the bottom half of Figure 1 – very big holes for brain tissue!
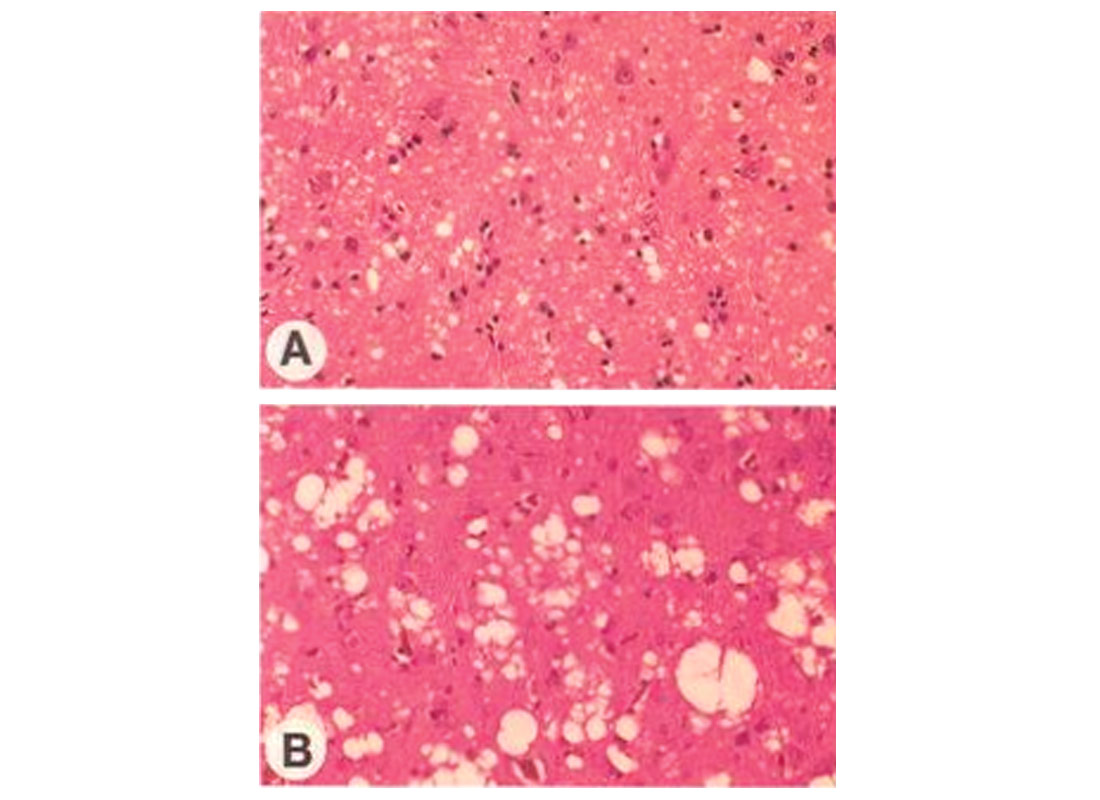
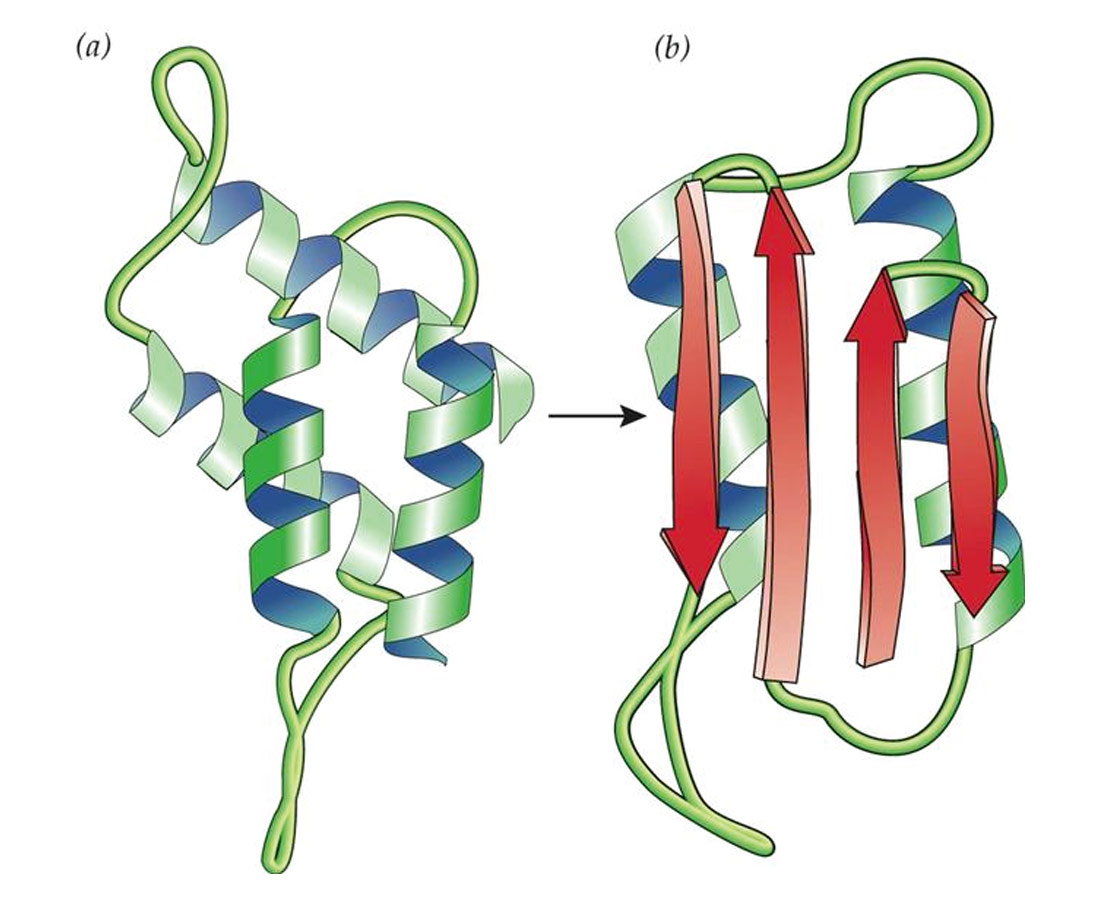
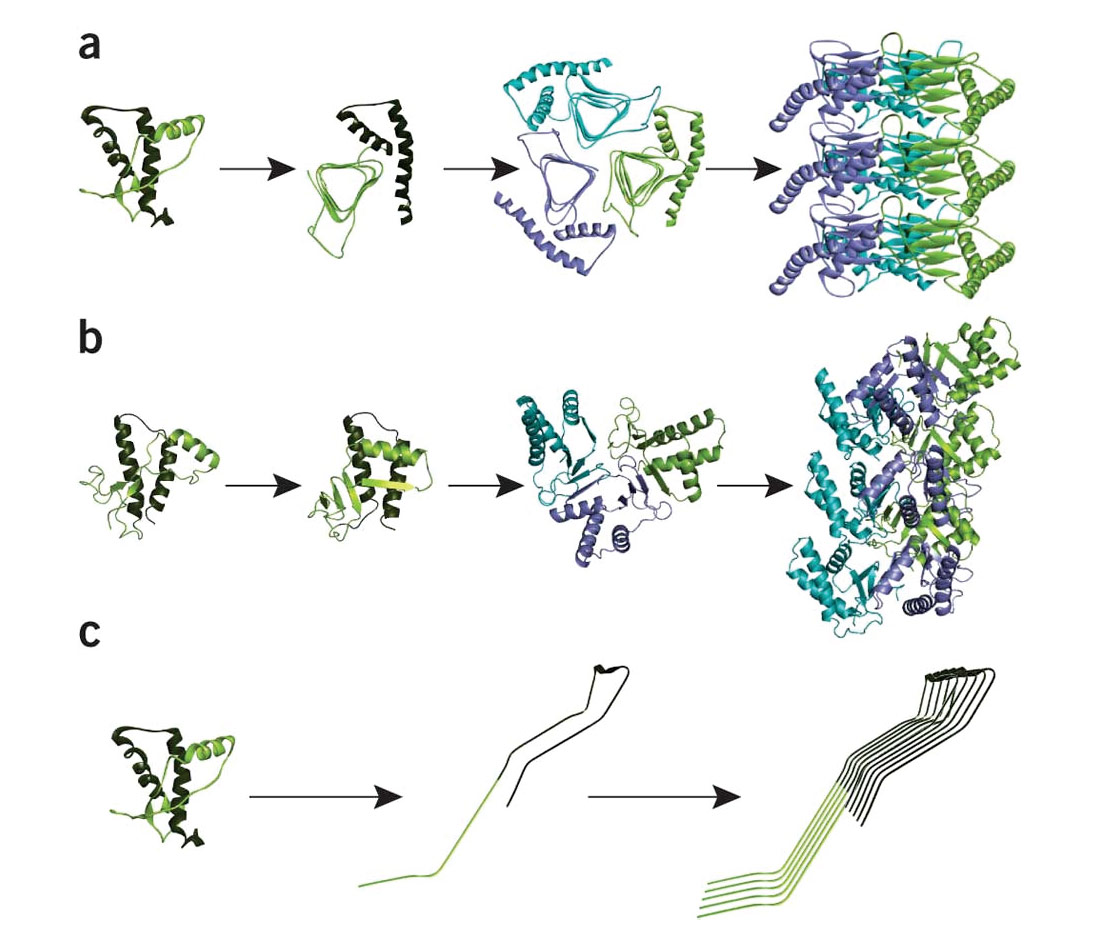
The actual biochemistry of what is going on is, quite simply, beyond my comprehension – it’s an area of science that is really terra incognito for me. Normally I find diagrams help me understand things, but the ones depicting the conversion of PrP into PrPSc don’t help me much, although they are pretty (Fig. 2). The conversion of two of the green helical structures into red sheet-like ones is obvious, and one can imagine the latter being pleated into nasty macramé-like fibrous clusters. There are a few theories explaining the actual change from the one form to the other, some involving additional actors which facilitate the change. Figure 3 graphically illustrates some of these theories. Again, one can see that the healthy PrPC has an independent, helical form, while the unhealthy PrPSc forms fibrous, sheeted clusters. The body does not have any enzymes that can break down the amyloid aggregates, so they keep growing if they can, and eventually cause cells to die.
Several of the theories explaining the conversion/replication process have fallen out of favour as they do not fully explain observed characteristics of BSE diseases, such as the long incubation period and rate of exponential growth in terms of PrPSc numbers. A currently popular theory posits that conversion happens when PrPSc fibrils are in contact with PrPC; normally the fibrils have two ends, but breakages can occur such that two ends become four, and then the extra ends latch onto the healthy prions and conversion occurs. Let’s just say the chemical properties of the amino acid arrangement within the PrPSc protein induce a rearrangement of the amino acids within the healthy PrPC such that the healthy proteins become replicas of the unhealthy ones, and become attached to them in an ever-growing aggregation. It’s kind of like a protein cult that converts more and more normal proteins over to its unhealthy cult structure (there, I just anthropomorphized the prion).
Transmission
Another reason that prion diseases command a high fear factor is their association with cannibalism. While in most cases prion diseases are transmitted via the ingestion of infected tissue, there are actually several ways the diseases can transmit. There are some prion diseases that are hereditary, but they are rare because they can only exist if (1), there is an heritable gene mutation that results in the creation of improper, i.e. PrPSc, prions, and (2) the genetic flaw only manifests itself later in life after the carrier has had a chance to reproduce and pass on the genetic flaw to his/her offspring. Fatal Familial Insomnia is a good example of such a disease, with only ~40 families identified worldwide as carrying the mutation.
Beyond the rare hereditary forms of TSE, transmission of prion diseases can happen via several mechanisms. For example, scrapie and chronic wasting disease transmission can apparently occur in several ways - via urine (infected sheep pees on grass, healthy sheep eats grass), in ewe-to-lamb milk, during birth (accidental ingestion by the lamb of placental tissue or amniotic fluid), and there is evidence that the scrapie prions can find their way into the soil (feces and urine) and remain active for several years. Clay rich soils bind better with the PrPSc prions than sandy soils, and sheep tend to eat clay-rich soil. That’s actually another interesting topic, the instinctive ingestion of clays by animals, apparently due to the tendency of clays to neutralize toxins. Amazonian macaws have evolved to eat clay, and this allows them to safely eat toxic seeds, a valuable source of nutrition off limits to competing species (Cornell University College of Agriculture and Life Sciences, 2014). There is evidence that Tanzanian chimps have learned to eat clay and Trichilia rubescens leaves, and this combination creates a “medicine” that fights the Plasmodium parasites that cause malaria (Norris, 2010). There is a tendency for women, especially in sub-Saharan Africa, to crave clay during pregnancy (The Week, 2011), something called geophagy. I have never encountered a woman who has admitted to such a craving, despite many years of discrete inquiry. Perhaps we just don’t have enough access to dirt in our over-sanitized world… but anyway, enough digression and back to prion transmission!
There have been a few flare ups of mad cow disease (BSE) in recent years, and the meat industry is terrified of it, as it is apparently able to make the leap over to humans, manifesting itself as new variant Creutzfeldt–Jakob disease (vCJD or nvCJD), similar to but not the same as Creutzfeldt–Jakob Disease (CJD). In Alberta we have seen the severe trade repercussions from just the hint of a BSE outbreak. It is believed that BSE is actually the bovine form of scrapie. The meat industry feeds stock animals something known as meat and bone meal (MBM, but when we eat it we call it hot dogs), and it is believed that MBM containing ground up tissue (including brain and spinal column tissue) from scrapie-infected sheep was fed to cows, and the cows are a suitable host as well.
The prion disease kuru has been conclusively linked to cannibalism. As far as I know, the only known outbreaks occurred in certain parts of Papua New Guinea in the first half of the 20th century, and petered out around 1970 along with the ritualistic cannibalism formerly practiced in these parts. The transmission mechanism appears simple. People with the disease (probably still in its incubative form) were killed and eaten, and then exacted a form of posthumous revenge, as their eaters would eventually experience a horrific death. Researchers believe that this 70 year or so long episode of the disease probably started with one individual who through a genetic mutation became afflicted with some variant of Creutzfeldt–Jakob Disease (CJD). He was killed and eaten, and then the prions sporadically found their way through the larger region of communities involved in the cannibalism. The incubation phase lasted up to 40 years, lots of time to roam around and be killed and eaten, and then the symptomatic phase about one year.
A little detail about kuru transmission for some reason really hit home with me. The males tended to get the best body parts during these cannibalistic rituals, with women and children getting the less desirable parts of the body, which included most importantly the brain, which is where most of the PrPSc would have been found. This meant that the majority of the deaths were among the women and children. I can imagine this would have been devastating for the communities, but when I find myself ruminating about that aspect I suddenly am reminded that these people were sitting around and chopping up and eating other human beings – the juxtaposition of these two forms of extreme human tragedy is just too bizarre to contemplate.
Kuru
I’ve already gone into the transmission of kuru in some detail. In 1960 the Australian administration in PNG imposed an outright ban on cannibalism. Thankfully with the decline and disappearance of cannibalism, kuru has become a thing of the past, although the incubation period of up to 40 years meant that isolated cases were still popping up in the early 2000’s. If you watch documentaries on PNG, they tend to push the cannibalism angle because it titillates a western audience in a macabre way; because of this, inevitably these documentaries dredge up some old guy who reluctantly admits that he can remember eating human flesh in his younger days.
In the Fore language spoken in the region of PNG where most of the cases occurred, the word kuru means ‘to shiver’. The damage caused by the kuru prions tends to occur in the cerebellum, which plays an important role in controlling movement, and so common symptoms include spasmodic shaking, trembling, slurred speech, and difficulty walking. The general medical term for this is ataxia, meaning ‘lack of order’, in other words the loss of conscious control of muscular movement. If you search for video clips with ‘kuru disease’ there are a number out there that clearly show the physical effects. Many victims also experienced difficulty chewing and swallowing, so their eventual deaths due to brain damage were hastened by malnutrition and starvation (Fig. 4).

Another unfortunate symptom was uncontrollable outbursts of unwarranted laughter; sometimes kuru has been called the ‘laughing disease’. The Fore people’s belief system explains many things with what we would call witchcraft or sorcery, and so rather than kuru being viewed as a medical issue, it was seen as the result of a curse being placed on the victim by someone else. The maniacal laughing undoubtedly made this easier to believe. These kinds of cultural factors can interfere in the treatment of infectious diseases, for example media reports say many Africans believe AIDS is spread by the condoms western doctors and charity workers distribute. In the case of kuru I don’t believe that kind of situation occurred – the eradication of cannibalism was going to happen regardless of whether kuru existed or not, simply because all outsiders to PNG found the practice so abhorrent, and there wasn’t the wealth or societal resilience to withstand the influences of the outside world – and so the removal of cannibalism took away the only effective vector for the disease.
Creutzfeldt-Jakob Disease
While rare, compared to other forms of TSE found in humans, CJD is quite common. The number of cases per year in the USA is about 300, and a similar rate of incidence would be expected in any other jurisdiction. Most (at least 85%) cases of CJD are identified as sporadic (sCJD) – in this form previously healthy prions spontaneously convert to the deadly form, and then the fatal chain reaction begins. Typically this occurs late in life, 60 years or older, after an extremely long incubation period of up to 50 years. Once this disease manifests itself, death usually occurs within a year. Up to 15% of cases are due to an inherited genetic mutation to the gene that carries the code for the prion protein (PRNP) and this form is called familial Creutzfeldt-Jakob disease (fCJD), and lastly a very small proportion of cases (<1%) is due to the physical introduction of infectious prions from a diseased person, usually through some type of transplant, such as corneal transplants, brain lining grafts and other procedures that introduce material likely to contain neural tissues and hence, prions.
There is a broad range of CJD symptoms, including rapid onset dementia, and ataxia, blindness, coma and many other nasty things soon follow. Really, the differences between kuru and CJD are not significant to the casual observer, although the difference in age of the afflicted is notable. However, that can be explained by the fact that the young were more likely to eat the diseased brains in the case of kuru. As with other TSE diseases, no effective cure has been found for CJD.
Bovine Spongiform Encephalopathy (BSE)
Cows suffering from BSE also show ataxic symptoms, as well as aggressive behaviour, anemia, and unusual in cows, antisocial behaviour; like kuru, the damage is high in the cerebellum. The incubation period varies between two-and-a-half and eight years, and once the disease expresses itself the cows die quite quickly. There are three basic theories relating to the origin of BSE – that it is a naturally occurring disease that has been present in cows since at least the Middle Ages, that it is actually the bovine form of scrapie, having jumped species only recently, and lastly that it is the result of a spontaneous genetic mutation much like sCJD is in humans. There is general consensus however that the recent BSE outbreaks have been caused by cattle being fed tainted MBM.
The problem (at least to us humans) is that quite often cows are killed and processed for their meat while still in the incubation phase, and then the risk is that humans will eat meat containing the diseased prions. As mentioned earlier, this can result in the disease jumping species, and manifesting itself in another human form of prion disease called new variant Creutzfeldt–Jakob disease (vCJD or nvCJD). There have been 229 known cases of nvCJD since it was first identified in 1996 as a unique form of TSE, and all involved people who had eaten beef and/or lived in or visited a country with a history of BSE outbreaks. One of the 229 cases may possibly be attributable to prion transfer via a blood transfusion. Besides this single possible case, there is overwhelming evidence that the only way for humans to get nvCJD is to eat diseased beef or beef products.
The symptoms and other aspects of nvCJD are very similar to classic CJD, with two main differences. First, the median age of death for CJD victims is in the late 60’s, while for nvCJD it is less than half that, in the late 20’s. CJD symptoms usually start with the classic ataxic signs of neurological damage, whereas with nvCJD the physical neurological symptoms are preceded by symptoms that are more psychological / behavioural in nature, by up to several months. But ultimately the outcome is the same for both – an unpleasant and untimely death.
Because of this demonstrated ability to jump over and afflict humans, since 1996 there has been an incredible focus on reducing or eliminating nvCJD and hence BSE, and there has been much research devoted to this. Efforts focus on detection – for example in Japan, all cows are tested prior to being slaughtered, and testing methods have become more and more reliable, efficient and cost effective; containment – most or all countries have implemented stringent regulations banning the feeding of MBM and other forms of feed containing body parts of other animals, especially ruminants, or at the very least identifying and banning parts of the body that are at risk to contain TSE prions; avoidance – beef consuming countries are now quick to impose bans on beef imports from countries that have experienced BSE outbreaks. One interesting fact that came out of all this is that it was demonstrated that unhealthy prions remain unaffected by high temperatures unless the temperature exceeds ~ 600 C, meaning that cooking meat is useless if you want to avoid being infected by TSE diseases.
Alternate Hypotheses
As in most fields, there are several areas of active debate. Many medical researchers do not accept what is known as the Prion Hypothesis because it has been gospel in biology for so long that replication is only possible if nucleic acids are involved. There are some reasons to doubt that healthy PrP prions are able to convert to unhealthy ones without any outside help.
First of all, there is clear evidence that certain genes play a role in some forms of TSE. At the very least this may suggest that a genetic predisposition may be required for TSE disease to take hold, and this is certainly the case with scrapie which requires both the introduction of PrPSc prions and the presence of a genetic susceptibility.
Second, researchers have been unable to simulate the conversion process, that is starting with healthy PrP, introducing PrPSc and then getting an exponential rate of conversion of the former to the latter has not been achieved. Also, it has been observed that the conversion tends to happen in specific parts of the brain, and to follow predictable patterns. All this suggests that some other factors are involved, and so there is much research into what is known as the “multi-component” or “cofactor variation” theory. Experiments in this area suggest that indeed other components are required for conversion, and these include various types of lipids, polyanionic molecules, and possibly RNA.
A very credible alternative to the Prion Hypothesis is that TSE’s are caused by some slow acting virus or viruses. Evidence supporting this includes the fact that viral material can be commonly found in TSE infected tissue (especially with scrapie), and the observation that TSE disease characteristics such as long incubation, fast onset, and symptomatic variations are all what you would expect with a virus. However, recent studies have poked holes in the virus theories, and so if an alternate theory is to replace the Prion Hypothesis, it is most likely to be the multifactor one.
This leads me to the “big question” that is asked around TSE – were prions somehow involved in the formation of life on Earth? It is certainly tempting to wonder whether the spontaneous replication of prions represents some vestigial step that occurred during the formative stages of life on Earth. One fact proponents of such an argument like to use is that the resistance of protein structures to high temperatures and toxic chemicals certainly makes them likely candidates given the presumably high temperatures of the Earth’s primordial chemical stew, which would probably severely damage modern RNA and DNA. Also, modern fungi contain prions that may have evolved to serve some useful purposes to the fungi. In my ignorance I have wondered if perhaps life’s replication framework was originally built around prions, and that later on the nucleic acids developed and evolved and eventually took over. This is an extremely interesting line of enquiry that I have not had time to pursue. However in writing this article I stumbled on an article which essentially confirmed that my ignorant speculations might not be so dumb after all – I will end with the following quote from that article.
Life may have started mainly on a protein related basis, before the modern genetic mechanisms of life forms known today started to develop, making DNA/RNA-based evolution more of a final step than the initial step of evolution. However, such a slow and rather inefficient type of life would have been eliminated quickly by the newly emergent RNA/DNA-based mechanism, and its existence would only be recognized nowadays as some weird epigenetically [sic] modulation in fungi and among the lethal spongiform encephalopathies in mammals. Maybe, prions represent the reminiscence of a very ancient analogical code of biological transmission of information rather that the digital one represented by modern nucleic acids
(Lupi, Dadalti, Cruz, & Sanberg, 2006).








Share This Column